Mopato - diagnoza poważnego. W tym artykule lekarz mówi o rodzajach tej wielostronnej choroby.
Treść
 «Jesteśmy trzema braciami (31 lat, 27 i 27 lat) - z dorastaniem cierpię na jedną chorobę - progresywną dystrofię mięśni. Wszystkie trzy - wyłączone. Może specjaliści zostaną spłacili na naszym nieszczęściem i pomocy.»
«Jesteśmy trzema braciami (31 lat, 27 i 27 lat) - z dorastaniem cierpię na jedną chorobę - progresywną dystrofię mięśni. Wszystkie trzy - wyłączone. Może specjaliści zostaną spłacili na naszym nieszczęściem i pomocy.»
«W dziecku (10 lat), progresywna dystrofia mięśniowa DECEDA. Lekarze bezsilni. Będę wdzięczny za wszelkie przepisy, wskazówki.»
«Wnuk (3 lata) nagle zaczął zaprzeczać nogami i z czasem się pogarsza. Lekarze umieszczają różne diagnozy i nic nie mogą robić. Pomóc dobrą radę.»
Są to fragmenty z liter ludzi, którzy zderzyli się z tak poważną chorobą jak mikopatia. Czym jest Myopatia? Spróbujmy sklasyfikować ten multi-wykres.
Myopatia i jego typy
Moopathy reprezentuje grupę chorób nerwowo-mięśniowych, które przejawiają się zmęczeniem, słabości mięśni, zmniejszenie tonu mięśniowego, zanik mięśni. Movathy, w zależności od czynnika przyczynowego, jest podzielony na progresywną dziedziczną dystrofię mięśniową, mikopatię hormonalną (choroby wewnętrznych gruczołów wydzielania) i miopatii metabolicznej (zaburzenia metaboliczne).
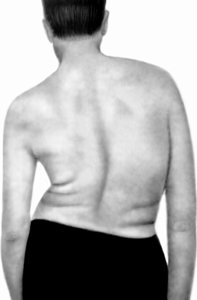
Porozmawiaj o postępowej dziedzicznej dystrofii mięśniowej. Ten rodzaj miopatii charakteryzuje się atrofią mięśniową ze względu na zniszczenie komórek mięśniowych ze względu na wadę specjalnego białka, który wzmacnia strukturę włókien mięśniowych. Białko to jest wytwarzane pod kontrolą specjalnego genu komórki, który znajduje się na 6. chromosomie ludzkim, a podczas wady tego genu pojawia się stopniowe zniszczenie skorup komórek mięśniowych, a następnie odlewnicze włókna mięśniowe.
Ten wadliwy gen jest dziedziczony, jeśli małżeństwo między krewnymi. Zmiany genów w 30% przypadków występuje w wyniku mutacji, czyli w tych przypadkach małżeństwo między krewnymi - ani. Choroba jest dziedziczona z prawdopodobieństwem 50%, jeśli jeden z rodziców dziecka jest chory. Jest wiąże się z chromosomem seksualnym kobietą i jest przekazywany, z reguły, synów, chociaż same kobiety mogą nie zranić. Zanikanie mięśni rąk ramienia, plecy, pasek miednicy i nogi.
W zależności od lokalizacji choroby, wiek, ciężkość chorób przeznaczy różne formy dystrofii mięśniowej. Tak więc forma młodzieżowa Erba Rota występuje w wieku 10-20 lat, gdy zanik mięśni pasa na ramię i ręce wydają się niezauważalnie, a następnie - pasek miednicy i nogi. Chodząc pacjentem, brzuch i skręcone tył klatki piersiowej. Wyróżnić się z położenia leżącego, pacjent odwraca się na boku i pochylając ręce na biodrach, stopniowo podnosi swoje ciało. Choroba powoli się rozwija.
Forma dzieci mięśniowej dystrofii dystrofii DECEDA zaczyna się w wieku 3-5 lat z zanikiem mięśni miednicy, biodra z jednoczesnym pogrubieniem mięśni oscraconalnych nogi (fałszywe pogrubienie). Stopniowo zanik mięśni pasów ramionowych i rąk. Dzieci początkowo zakłócały chód, a następnie powstają trudności w ruchu. Wielu ma tętno ze względu na wzrost rozmiaru serca. Progresja choroby lub jego złośliwy przepływ z powodu wczesnego unieruchomienia kończyn prowadzi do smutnego wyniku. Są chory, głównie chłopcy (1 za 3000). Być bardziej dokładnym, mężczyźni i kobiety są również chorzy. Tylko choroba Dosene manifestuje się w chłopcach. Dziewczyny są nosicielami tego genu.
Ale zdarza się i łagodne dla dystrofii mięśniowej (Becker Miodastrophy), kiedy choroba manifestuje się powoli, zwłaszcza w małych dzieciach. Przez wiele lat zachowują zadowalającą kondycję fizyczną i tylko przystąpienie różnych chorób ostrych i urazów prowadzi ich do unieruchomienia, wyczerpania z złym wynikiem.
Postać miarectora na ramię Miodyatrofii, zwana Landuzy-Dezhard, która może być w wieku od 6 do 52 lat (częściej w ciągu 10-15 lat) i charakteryzuje się klęską mięśni twarzy ze stopniową późniejszą atrofią Mięśnie pasa naramiennego, tułowia i kończyn. Wczesne objawy choroby są słabo zamknięte i nieuzasadnione powieki, w pełni zamknięte usta, które tworzą rozmytą mowę i niemożliwość policzków. Choroba zachęca powoli. Przez długi czas pacjent może poruszać się i utrzymać zdolność do pracy, a następnie po 15-25 latach mięśni pasa miednicy nóg są stopniowo zbrodniane, co utrudnia poruszanie się.
Przydzielono również grupę wtórnej stopniowej dystrofii mięśniowej, która powstaje w związku z uszkodzeniem nerwów: Neuronowa, kręgosłupa Miodastrophia, zwana nadal amyotrofią.
Obraz Shark-Marie Amyotrofy, który charakteryzuje się stopniową zanikiem małych mięśni zatrzymania, są następnie zanikiem mięśni nóg i dolnej części bioder, a mięśnie środkowych i górnych części bioder nie Zmiana i uda jest kształtem butelki z szyją, przechyloną. Mięśnie rąk i przedramionami są następnie stopniowo zanikiem. Mięśnie tułowia, pasek na ramię i twarz. Choroba występuje w wieku 18-25 lat, powoli postępuje i stabilizuje.
Wrodzona zanik mięśniowego kręgosłupa Kugelberga-Vallager charakteryzuje się stopniową zanikiem mięśni rąk, nóg, opóźnienia rozwoju psychicznego i fizycznego, deformacji kręgosłupa. Choroba objawia się w wieku 8-10 lat i powoli się rozwija.
Progresywny Ayotrofony Aran-Duzhen rozpoczyna się w wieku 25-50 lat i objawia atrofię mięśni pędzli. Następnie reszta rąk jest stopniowo zblakowana, potem stóp ciała, w t.C. mięśnie międzypostalnymi, co powoduje zaburzenia oddechowe, z których przychodzi śmierć.
Wrodzona Amiotonia (zmniejszenie odcienia mięśni) Oppenheim charakteryzuje się słabością mięśni z powodu ich niedostatności, a dystrofia mięśniowa jest drugorzędna. W noworodka nie rozwija się, ale wnikanie infekcji dróg oddechowych może powodować zapalenie, a śmierć przychodzi w pierwszym roku życia. Z wieku, funkcja silnika mięśniowego poprawia.
Leczenie dystrofii mięśniowej ma na celu spowolnienie procesów dystroficznych (niszczenia) w mięśniach, a nawet ich wypowiedzi. Jednak radykalne traktowanie nie zostało jeszcze znalezione. Chociaż nadzieja jest na terapii Genin, która zaczyna powoli wdrażać w praktyce medycznej.